Спинальная мышечная атрофия (СМА), или спинальная амиотрофия Верднига-Гоффмана является аутосомно-рецессивным наследственным заболеванием, которое характеризуется прогрессирующей гипотонией и мышечной слабостью.
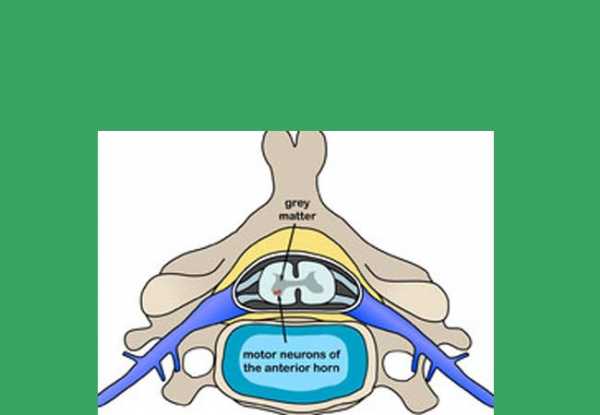
Характерное ослабление мышечных тканей происходит из-за прогрессирующей дегенерации альфа двигательных нейронов в передних рогах спинного мозга. Таким образом, в основе болезни заложена патология спинного мозга, которая способна передаваться по наследству.
Особенностью заболевания является более активное проявление слабости в скелетных мышцах, расположенных более глубоко, чем тех, которые расположены ближе к поверхности тела. В этом материале мы расскажем о симптомах и лечении спинальной мышечной атрофии Верднига-Гофмана.
Информация будет полезна всем, кому по ряду причин пришлось столкнуться с этим серьезным часто – смертельным заболеванием.
У некоторых пациентов в патологический процесс также могут быть вовлечены моторные нейроны черепно-мозговых нервов, особенно с V по XII пару. В этом случае болезнь берет свое начало от заднего рога клеток спинного мозга, что в дополнение ко всему обуславливает недостаточность мышц диафрагмы, желудочно-кишечного тракта, сердца и сфинктеров.
В 1890 году Вердниг впервые описал классическую инфантильную форму СМА – проявление синдрома у детей раннего возраста. Много лет спустя, в 1956 году, Кугельберг и Веландер классифицировали менее тяжелую форму спинальной мышечной атрофии у более старших пациентов.
Благодаря этим ученым, сегодня врачи могут точно дифференцировать СМА от разных типов сходных по симптомам болезней, например, мышечной дистрофии Дюшенна.
Спинальная амиотрофия является наиболее распространенным диагнозом у девочек, причем с сильно прогрессирующей слабостью. Это одна из наиболее распространенных генетических причин смерти у детей.
Синдром СМА делится на четыре типа на основе возраста пациента следующим образом:

Частота возникновения болезни составляет примерно один случай на 15-20 тысяч человек. Если говорить только о новорожденных, то этот показатель составит примерно 5-7 случаев на 100 тысяч. Поскольку спинальная амиотрофия является наследственным рецессивным заболеванием, многие родители могут быть его носителями и не знать об этом.
Распространенность лиц-носителей СМА – 1 из 80, другими словами у каждой 80-й семьи может родиться ребенок, страдающий спинальной мышечной атрофией. Такой риск возрастает в несколько раз, когда оба родителя являются носителями мутантного гена.
Таким образом, синдром СМА – наиболее распространенное дегенеративное заболевание нервной системы у детей после муковисцидоза и, как уже отмечалось выше, это – ведущая наследственная причина детской смертности.
Летальный исход обусловлен дыхательной недостаточностью. Чем моложе пациент на ранних стадиях болезни, тем хуже прогноз. Общий средний возраст на момент гибели составляет около 10 лет. Состояние интеллекта и других показателей психосоматического развития ребенка никак не влияет на прогрессирование заболевания.
В отличие от маленьких пациентов, взрослые мужчины чаще поражены болезнью, чем женщины примерно в соотношении 2:1 при том клинический курс у пациентов мужского пола является более суровым. Учащение случаев заболевания у женского пола начинается примерно с 8 лет и мальчики «догоняют» девочек на уровне 13-летнего возраста.
Первый тип спинальной мышечной атрофии обуславливает появление первых симптомов еще до рождения ребенка. Большинство матерей сообщают о ненормальной неактивности плода на поздних стадиях беременности. Симптомы СМА у новорожденных проявляются достаточно явно – ребенок не может самостоятельно перевернуться, а в последующем занимать сидячее положение.
Кроме того, развивается прогрессивное клиническое ухудшение, которое в преобладающем большинстве случаев завершается летальным исходом. Смерть обычно наступает от дыхательной недостаточности и ее осложнений у пациентов в 2-х лет.
Пациенты со вторым типом СМА нормально развиваются в течение первых 4-6 месяцев жизни. Они могут быть в состоянии сидеть самостоятельно, но они никогда не смогут ходить, в будущем им потребуется инвалидная коляска для передвижения. Как правило, такие дети живут гораздо дольше, чем пациенты, страдающие спинальной атрофией Верднига-Гоффмана. Средний срок жизни – до 40 лет.

Пациенты с третьим типом болезни часто с трудом поднимаются по лестнице или встают с пола, в первую очередь по причине слабости разгибателей тазобедренного сустава. Продолжительность жизни близка к нормальной.
Новорожденные с первым типом спинальной мышечной атрофии неактивны. Они с огромным трудом двигают конечностями, если вообще на это способны. Бедра почти постоянно в согнутом состоянии, ослаблены и их легко можно выкручивать руками в разные стороны. Колени также согнуты.
Поскольку внешняя мускулатура обычно менее поражена – пальцы рук и ног двигаются почти нормально. Младенцы не могут контролировать или поднимать голову. Арефлексия (отсутствие рефлексов) наблюдается почти у всех больных.
Дети, страдающие вторым типом СМА способны двигать головой и 75% этих пациентов могут сидеть самостоятельно. Мышечная слабость сильнее в нижних конечностях, чем в верхних. Рефлекс коленной чашечки отсутствует. Более старшие дети могут продемонстрировать рефлексы двухголовой мышцы и трицепсов.
Сколиоз является наиболее частым симптомом СМА, а также у большинства пациентов развивается вывих бедра, одно- или двусторонний. Указанные признаки развиваются в возрасте моложе 10 лет.
Пациенты с третьим типом болезни спинальной мышечной атрофии могут ходить в начале жизни и можно поддерживать эту способность амбулаторно на протяжении всего подросткового возраста. Слабость может привести к падению стопы, а также ограниченной выносливости организма. Треть пациентов становятся прикованными к инвалидному креслу по достижении возраста в 40 лет.
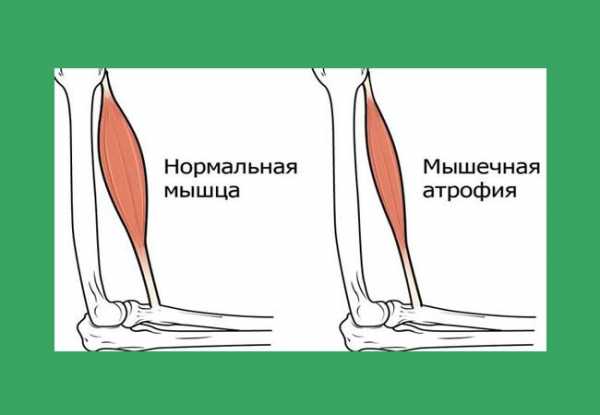
В настоящее время не существует никакого известного медицинского лечения спинальной мышечной атрофии, поэтому сразу стоит отметить, что процент выживаемости среди пациентов раннего и среднего возраста достаточно низок.
По причине низкой продолжительности жизни, новорожденные со спинальной мышечной атрофией Верднига-Гоффмана, из-за их короткой продолжительности жизни, требуют малого участия со стороны ортопеда. Шинирование используется в случае переломов, что нередко наблюдается при ослабленной мышечной активности.
Для пациентов II и III типа СМА, физическая терапия может быть использована для лечения контрактуры суставов (ограниченного их движения). Для более радикального лечения контрактуры показано хирургическое лечение.
Как уже отмечалось выше, наиболее распространенной ортопедической проблемой при синдроме СМА является сколиоз, который часто принимает тяжелую форму. Прогрессия кривизны позвоночника составляет около 8° в год, несмотря на лечение брекетами.
Задний спондилодез сегментарного типа часто рекомендован для молодых пациентов, у которых кривизна позвоночного столба не может быть исправлен фигурными скобами, а также для пациентов старше 10 лет с кривизной более 40°.
Хирургическая операция должна быть задержана до тех пор, пока медицинской точки зрения это возможно. Следует иметь в виду, что прогрессирование кривизны происходит медленнее у пациентов с третьим типом СМА и появляется чаще в более позднем возрасте.

Лечение спинальной мышечной атрофии требует индивидуального подхода к нормированию меню пациента, что, к сожалению, очень часто не соблюдается лечащими врачами. Антропометрические показатели, состав крови и биохимические маркеры состояния мышц являются важными элементами оценки у пациентов с СМА.
Особенность течения болезни у конкретного пациента может потребовать вмешательства в его рацион, чтобы повлиять на выше перечисленные показатели, поскольку именно с помощью продуктов питания мышцам можно дать те питательные вещества, которые необходимы пациенту в его случае.
Разумеется, такой подход к лечению спинально-мышечной амиотрофии актуален лишь при постановке диагноза на вторую или третью форму болезни.
Дополнительная поддержка пациента, страдающего спинальной мышечной атрофией второго и третьего типа актуальна не меньше, чем диета. В первую очередь с помощью нормированной нагрузки можно предотвратить прогрессирование контрактуры суставов, а также поддержать силу, выносливость и независимость в самообслуживании.
Достаточно весомую роль физические упражнения играют в образовательной, социальной, психологической и профессиональной деятельности пациента, поскольку у него появится возможность вести практически нормальный образ жизни, как и здоровые люди.
zdravo-bravo.ru
SMN-связанная СМА, или 5qSMA, или проксимальная СМА обычно подразделяется на три категории. Условно можно выделить еще СМА0 (ноль) и СМА4. Таким образом, есть несколько основных типов СМА, и все они протекают по-разному. Процесс может развиваться в различные периоды жизни, иметь свои клинические особенности, свой характер течения, прогноз и необходимый объем помощи и поддержки.
Тип 1 – наиболее тяжелый, с самым ранним дебютом, тип 3 – наименее тяжелый, с поздним возрастом начала. Некоторые специалисты выделяют еще тип 4 для обозначения умеренной или мягкой СМА с дебютом во взрослом возрасте.
Специфика заболевания в том, что у каждого ребенка, даже в пределах одной группы, СМА протекает по-разному, индивидуально. Внешне это проявляется в возможном объеме движений – некоторые дети способны удерживать голову, немного поднимать руки и приподнимать ноги, а другие просто лежат в классической позе «лягушка» и могут только чуть-чуть шевелить стопами и пальчиками кистей рук.
Виктор Дубовиц, один из корифеев в области СМА-ведения, в 90-е годы предложил помимо обычной классификации использовать более усложненную шкалу. Например, есть СМА1 и ее подтипы будут 1.1, 1.2, 1.3, т.е. от 1.1 до 1.9. Такая схема используется в Италии.

Виктор Дубовиц. Неразбериха в классификации СМА: возможность решения
Американская система основывается на шкале ABC, когда В – это классика, А – слабый тип, а С, соответственно, более сильный. Система с подтипами ABC наглядно описывает клинику СМА и позволяет подбирать для ребенка поддерживающую терапию в зависимости от подгруппы и соответствующих прогнозов. Этой системой начинают использоваться в России и других странах.
Важно! С помощью генетического теста тип СМА не определяется. Тип устанавливается на основе функциональных возможностей ребенка.
Симптоматика заболевания проявляется внутриутробно в отсутствии у плода двигательной активности. С рождения у ребенка выражена генерализованная мышечная гипотония с характерной позой «лягушки», резко снижена спонтанная двигательная активность. Сухожильные рефлексы не вызываются.
Как правило, эти дети долго наблюдаются педиатрами и неврологами с диагнозом перинатальная энцефалопатия. Иногда врачи связывают симптомокомплекс вялого ребенка с тяжелыми родами. Но все дети с перинатальной энцефалопатией и с последствиями тяжелых родов достаточно быстро и хорошо адаптируются, постепенно улучшаются, в отличие от детей со СМА.
Прогноз крайне неблагоприятный – дети погибают, как правило, в очень раннем возрасте (до шести месяцев) от интеркуррентных заболеваний (осложняющих течение основной болезни).
Обычно СМА0 и СМА1 объединяют.
При I типе спинальной мышечной атрофии (тип Верднига-Гофмана) мать уже во время беременности может обратить внимание на позднее и слабое шевеление плода. С рождения у ребенка выражено распространенное снижение мышечного тонуса (синдром «вялого ребенка»). С первых месяцев жизни появляются слабость и атрофия мышц верхних и нижних конечностей, с последующим вовлечением мышц туловища и шеи. Такие изменения мышц приводят к тому, что дети не могут сидеть. Мышечные атрофии и подергивания мышечных волокон обычно маскируются хорошо выраженной подкожно-жировой клетчаткой. Характерным симптомом является мелкое дрожание (тремор) пальцев вытянутых ручек. Иногда обнаруживаются подергивания мышц языка.
Типичным признаком также является ослабление или полное исчезновение сухожильных рефлексов (коленный, ахиллов), ограничение нормальной подвижности в суставе, деформации скелета. Вследствие слабости межреберных мышц грудная клетка ребенка становится уплощенной. Так как в результате слабости мышц не происходит достаточной вентиляции легких, присоединяются частые респираторные инфекции, возникают разнообразные дыхательные расстройства. Психическое развитие детей не страдает.
У младенцев могут возникать дыхательные нарушения и невозможность приема пищи. Врожденные контрактуры, варьирующие по степени выраженности от простой косолапости до генерализованного артрогрипоза (множественная врожденная патология аппарата движения, проявляющаяся многочисленными контрактурами суставов, мышечной гипотрофией и поражением спинного мозга), возникают примерно у 10 % новорожденных с тяжелым поражением. Дети грудного возраста лежат в расслабленной позе «лягушка», спонтанная двигательная активность снижена, дети не могут преодолеть силу тяжести конечностей, плохо удерживают голову.
Обычно, за исключением очень редких, тяжелых случаев, диагноз СМА не ставится в роддоме. Ребенка выписывают домой здоровым, и когда родители замечают низкий мышечный тонус, они или не придают этому серьезного значения, если не знают, как должен двигаться здоровый малыш, или успокаиваются советами доктора в поликлинике: «Не переживайте, все развиваются в свой срок, еще встанет и побежит». Родители могут не понимать всей тяжести проблем, это должен увидеть врач, но, к сожалению, в поликлиниках симптоматику СМА знают плохо.Эти дети проявляют первые признаки заболевания в возрасте до 6 месяцев. Это значит, что они не приобретают навыки самостоятельно сидеть, ползать, ходить. В итоге, объем движений у таких детей очень мал. При этом СМА не затрагивает когнитивную сферу, дети все понимают, не затронута и чувствительность. Если с ними заниматься, как с обычными детьми – играть, читать, собирать пирамидки, – то эти дети в умственном и психическом плане развиваются абсолютно нормально, и все задержки развития, которые могут им ставить неврологи, связаны с нарушением двигательной активности и условиями, в которых они находятся.
Более 2/3 детей с этим заболеванием умирают до 2-летнего возраста, во многих случаях смерть наступает в раннем младенческом возрасте в связи с поражением дыхательной мускулатуры и возникновением разнообразных осложнений со стороны легких.
При спинальной мышечной атрофии II типа заболевание впервые проявляется несколько позже (в первые 1,5 года жизни ребенка) и характеризуется более медленным течением. Главным признаком является невозможность ребенка встать на ноги.
Дети со СМА2 обычно способны сосать и глотать, дыхательная функция в раннем младенческом возрасте не нарушена. Носовой оттенок голоса и нарушение глотания появляются в более старшем возрасте. Несмотря на прогрессирующую мышечную слабость, многие из них доживают до школьного и более старшего возраста, хотя на поздних стадиях заболевания отмечается тяжелая степень инвалидизации, и дети нуждаются в инвалидных колясках. Обычные коляски им не подходят, необходимы дополнительные поддержки, упоры, специальные приспособления, оптимально фиксирующие положение тела.
У многих пациентов с большой продолжительностью жизни одними из основных осложнений заболевания становятся сколиоз и контрактуры, которые развиваются очень быстро. Даже у лежачих детей сколиоз развивается достаточно существенно, искривление позвоночника происходит не от нагрузки, а от слабости.
Дети со СМА2 некоторый период времени бывают достаточно стабильны, и родители могут поддерживать тот набор навыков, который этими детьми уже приобретен.
Есть такой термин «плато заболевания», который обозначает некий период, в течение которого дети достаточно стабильны, и значительного ухудшения состояния, прогрессирования болезни не происходит. Это может выглядеть так: дети набирают навыки, как все обычные дети, при «запуске» болезни перестают развиваться, а потом регресс состояния у них может идти очень медленно или, наоборот, очень быстро, у каждого ребенка по-своему. Или так: дети определенное время набирают навыки, потом происходит какое-то событие или болезнь, часть навыков теряется, и дальше наступает довольно долгое «плато», в течение которого могут быть даже незначительные улучшения, но потом наступают неизбежные ухудшения. Скорость прогрессирования болезни, продолжительность «плато» (или его отсутствие) и последующих ухудшений – все это очень индивидуально.
Болезнь Кугельберга-Веландер – наиболее легкая форма спинальной мышечной атрофии (СМА III типа). Начинается она в возрасте от 1,5 до 17 лет. С такой болезнью люди живут дольше, прогрессия идет медленно. Атрофия мышц начинается с ног и далее распространяется на руки. В младенческом возрасте клинические проявления заболевания могут отсутствовать. Прогрессирующая слабость развивается в проксимальных отделах конечностей, особенно в мышцах плечевого пояса. Пациенты сохраняют способность к самостоятельной ходьбе. Симптомы слабости мышц бульбарной группы появляются редко. Примерно у 25 % пациентов с этой формой СМА в большей степени выражена мышечная гипертрофия, а не атрофия мышц; поэтому может быть ошибочно диагностирована мышечная дистрофия. Пациенты могут дожить до зрелого возраста.
СМА у этой большой группы детей выявляется в возрасте старше полутора лет, т.е. обычно ребенок может самостоятельно ходить. Болезнь проявляется настолько индивидуально, что диагноз могут поставить в полтора года, а могут и в девять лет. В зависимости от этого будут разные прогнозы и по продолжительности, и по качеству жизни.
У детей со СМА3 продолжительность жизни почти не отличается от стандартной, они доживают и до 30, и до 40 лет. У них не так быстро прогрессируют все симптомы, но с этими детьми тоже надо проводить и реабилитационные занятия, и занятий по физической терапии.
Сейчас есть достаточно большое количество взрослых пациентов со СМА, и это не четвертый тип, это дети со СМА2 и СМА3, которые выросли. У них возникают большие проблемы, которые не связаны с перемещением и дыханием. Сколько бы они ни жили, 20 лет или 30, они уже разочаровались в медицине, потому что никто не знает, что с ними делать. Каждая мама ребенка со СМА и каждый взрослый пациент со СМА может рассказать примерно одну и ту же историю про прием у доктора: «Не приходите ко мне, мы не знаем, что с вами делать»; «Ты еще не умер, ах, ничего себе». Действительно, врачи не знают, что делать с этими пациентами, и говорят: «Ну, не лечится же, что ты хочешь от меня, правда, по-честному, я не знаю, что с тобой делать». Ими никто не занимается, после 18 лет вообще не знают, что с ними делать.
Эти пациенты (часто невидимые пациенты, так как сидят дома) не верят в медицинскую помощь, потому что всю жизнь от них отмахивались. И они обращаются за помощью только тогда, когда уже невозможно игнорировать симптоматику, когда сильная боль, т.е. поздно – практически уже на смертном одре. До недавнего времени с этой категорией пациентов не было никакой работы. Сейчас появилась возможность как-то улучшать ситуацию, больше помощи оказывают благотворительные фонды, этой проблеме уделяется больше внимания.
Заболевание СМА3 развивается не очень стремительно, постепенно происходит общее ослабление организма и медленная потеря приобретенных навыков.
Четвертый тип СМА проявляется у людей старше 25 лет. Заболевание развивается довольно медленно, практически не влияя на продолжительность жизни. При СМА4 может развиваться тремор, возникать общее ослабление, в том числе, и мышечной силы. Со временем СМА4 может приводить к потере способности к самостоятельному передвижению.
spravka.neinvalid.ru
14′17Авг
Одним из наиболее частых аутосомно-рецессивных заболеваний является спинальная мышечная атрофия (СМА).Спинальная мышечная атрофия – наследственное нервно-мышечное заболевание, причиной которого является постепенное и необратимое нарушение функций двигательных нейронов спинного мозга, приводящее к симметричному ослабеванию, а затем атрофии мышц. Хотя это заболевание относится к редким (орфанным), оно является вторым (после муковисцидоза) по распространенности из наследственных среди коренных европейцев и населения Северной Америки европейского происхождения.
Различают четыре типа СМА, в зависимости от возраста манифистации и степени проявления симптомов:
СМА 1 типа (младенческая форма), или болезнь Верднига-Гоффмана – самая ранняя и тяжелая форма заболевания. Обычный возраст начала 0-6 месяцев. Чаще всего дети с такой формой заболевания не доживают до 2-х лет.СМА 2 типа (промежуточная форма), или болезнь Дубовица – возраст проявления этой формы заболевания 6-18 месяцев. Дети с таким типом СМА могут сидеть без поддержки, но не могут вставать и ходить. Тяжесть заболевания и прогноз зависит от скорости вовлечения в болезнь мышц, отвечающих за дыхание.СМА 3 типа (ювенильная форма), болезнь Кюгельберга-Веландер – проявляется в возрасте старше 18 месяцев. Дети с этой формой СМА способны сравнительно длительное время ходить самостоятельно, слабость и атрофия мышц прогрессируют медленно.СМА 4 типа (взрослая форма) — самая легкая форма заболевания, обычно проявляющаяся на втором или третьем десятилетии жизни симптомами, аналогичными ювенильной форме. Как правило, не приводит к снижению продолжительности жизни пациента.
Генетической причиной СМА являются мутации в гене SMN1. Ген SMN1 кодирует белок, необходимый для работы двигательных нейронов. Важной частью этого белка является фрагмент, информация о котором содержится в 7 экзоне (определенной части) гена SMN1. Любая мутация, которая приводит к отсутствию этого фрагмента в белке или же значительно нарушает строение белка, может привести к развитию заболевания. Полное отсутствие 7 экзона (делеция) является самой частой причиной СМА. Если на обеих хромосомах в гене SMN1 есть нарушения в 7 экзоне, то нормальный белок не будет формироваться, а отсутствие нормально функционирующего белка вызывает СМА.
В геноме человека также есть ген SMN2 . Он очень похож на ген SMN1 (99% совпадения последовательностей). Одно из ключевых отличий этих двух генов – определенный нуклеотид в 7 экзоне. Одна замена в последовательности гена SMN2 приводит к тому, что с него синтезируется нефункциональный белок. Однако, в результате ошибок синтеза белка, которые иногда случаются в нашем организме, с гена SMN2 может синтезироваться небольшое количество правильного, функционального белка, необходимого для работы двигательных нейронов. Именно поэтому даже при полном отсутствии нормального белка с гена SMN1 количество копий гена SMN2 влияет на тяжесть симптомов.
Информация о количестве копий генов SMN1 и SMN2 позволяет диагностировать заболевание и оценить риск рождения в семье ребенка с диагнозом СМА. Если оба родителя являются носителями мутации в гене SMN1, вероятность рождения ребенка с СМА равна 25%. Однако обычными методами невозможно уточнить количество копий генов SMN1 и SMN2.
Правильная молекулярная диагностика носительства СМА и самого заболевания осложнена схожестью генов SMN1 и SMN2. Для диагностики необходимо использовать методы, позволяющие различить эти гены по немногим отличающимся точкам. Так как ключевую роль играет именно экзон 7, необходимо использовать метод, который позволяет определить количество его копий в гене SMN1.
Сегодня единственным методом точного определения количества копий каждого гена является анализ методом MLPA (мультиплексная амплификация лигазно- связанных проб). В результате анализа лаборатория получает заключение о количестве 7 и 8 экзонов генов SMN1 и SMN2. Все остальные методы позволяют выявить наличие/отсутствие указанных экзонов генов, но не определить количество копий каждого. Хотя у большинства пациентов со СМА выявляются делеции 7 и 8 экзона в гене SMN1, для молекулярного подтверждения диагноза ориентируются только на данные по количеству копий 7 экзона.
Методов эффективного лечения СМА не существует. В настоящее время 2 фармакологические компании разработали генно-терапевтические препараты для лечения СМА. Препарат SPINRAZA (Biogen) разрешен к использованию в ЕС, однако стоимость такого лечения очень высока. Препарат AVXS-101 (AveXis) находится на стадии клинических испытаний и когда он будет доступен для лечения детей со СМА неизвестно. Сейчас доступна только поддерживающая терапия, в зависимости от тяжести заболевания.
Поэтому так важно иметь возможность снизить риск рождения больного ребенка в семье. Для этого необходимо проводить генотипирование будущих родителей, которое позволит оценить риск для потомства. В случае выявления статуса носительства мутаций в гене SMN1 обоими родителями, им может быть рекомендована пренатальная или преимплантационная генетическая диагностика СМА.
Автор: Анна Орлова
биолог-генетик
genetico.ru
ЧТО ТАКОЕ - СПИНАЛЬНАЯ МЫШЕЧНАЯ АТРОФИЯ? Спинальная мышечная атрофия (СМА) — генетическое заболевание, поражающее область нервной системы, отвечающей за контроль движений произвольных (скелетных) мышц
Большинство нервных клеток, контролирующих мышцы, находятся в спинном мозге, отсюда — слово «спинальная» в названии болезни. Слово «мышечная» говорит о том, что страдают мышцы, которые не получают сигналов от этих нервных клеток. Ну и «атрофия» - медицинский термин для истощения или «усыхания», которое происходит с мышцами, когда они неактивны
При СМА происходит утрата нервных клеток в спинном мозге, именуемых мотонейронами, поэтому она классифицируется как болезнь мотонейронов
Возраст начала и тяжесть течения СМА варьируется у разных людей в широком диапазоне
Что является причиной СМА?
В большинство случаев СМА обусловлена дефицитом протеина мотонейронов, именуемым SMN (протеин выживаемости мотонейронов)
Этот протеин, как следует из его названия, необходим для нормального функционирования мотонейронов. Недавние исследования наводят также на мысль о том, что отсутствие SMN может также непосредственно влиять на мышечные клетки
Существуют также формы СМА, не связанные с протеином SMN
КАКИЕ ЕСТЬ ФОРМЫ СМА?
SMN-связанная СМА
SMN – связанная СМА обычно подразделяется на 3 категории. Тип 1 — наиболее тяжелый, с самым ранним началом, а тип 3 — наименее тяжелый, с наиболее поздним возрастом начала. Некоторые специалисты выделяют еще тип 4 для обозначения умеренной или мягкой СМА с дебютом во взрослом возрасте
Все эти типы связаны с генетическими поломками (мутациями) на хромосоме 5, что влияет на количество синтезируемого SMN – протеина. Более высокие уровни протеина снижают тяжесть СМА. Более подробно о том, как эти мутации приводят к развитию СМА, описано в разделе «Это семейное?»
Не связанная с SMN СМА
Существуют также формы СМА, не связанные с SMN и не являющиеся результатом мутаций на хромосоме 5. Подробнее об этом — в разделе «Что происходит с людьми с различными формами СМА?»
Спино-бульбарная мышечная атрофия (СБМА)
Этот тип СМА, именуемый также болезнью Кеннеди, является результатом мутаций гена на Х-хромосоме, и значительно отличается от SMN-связанных типов СМА. Более подробно — в соответствующем разделе.
ЧТО ПРОИСХОДИТ С ЛЮДЬМИ, ИМЕЮЩИМИ SMN-СВЯЗАННЫЕ ФОРМЫ СМА?
Тяжесть SMN-связанных СМА зависит от того, насколько рано проявляются симптомы, что в свою очередь взаимосвязано с количеством SMN протеина в мотонейронах. Более позднее появление симптомов и более высокие уровни SMN протеина предполагают, что течение заболевание будет более мягким
Однако в настоящее время большинство специалистов сомневаются в столь строгой зависимости и предпочитают не делать однозначных прогнозов по тяжести течения и продолжительности жизни, основываясь только на возрасте дебюта. Последние исследования говорят в поддержку такой гибкости
СМА тип 1 (болезнь Верднига-Гоффмана)
Если дети с СМА очень слабы в первые месяцы жизни и испытывают трудности с дыханием, сосанием и глотанием, по всей видимости трудно рассчитывать для них на хороший прогноз. Ранее считалось, что такие дети не выживают более двух лет. И в настоящее время в большинстве случаев такой прогноз оказывается справедливым
Тем не менее с применением технологий взамен естественных функций дыхания и питания такие дети могут выживать в течение нескольких лет. Механическая вентиляция легких (сейчас такие аппараты стали портативными, в отличие от применявшихся в предыдущие годы «железных легких» и тяжелых механизмов) и питание через зонды непосредственно в желудок, а не в глотку, могут продлить жизнь
Умственное и эмоциональное развитие, а также чувствительность при СМА совершенно нормальны
СМА тип 2 (промежуточная СМА)
Диагноз СМА тип 2 позволяет родителям и детям планировать будущее, несмотря на сокращенную продолжительность жизни. Этот тип СМА дебютирует в детском возрасте, но как правило позже младенчества. В некоторых источниках к типу 2 относят заболевание с возрастом дебюта в промежутке между 6 и 12 месяцами. В других источниках говорится, что заболевание классифицируется как тип 2, если ребенок способен сидеть без поддержки, если его посадить
При этом типе СМА мышцы, расположенные ближе к центру туловища (т.н. проксимальные мышцы) обычно поражены в большей степени, или по крайней мере поражаются раньше, чем мышцы, более удаленные от центра (дистальные). Например, мышцы бедер слабее, чем мышцы голеней и стоп.
Кроме того, ноги обычно ослабевают раньше, чем руки. Руки в итоге ослабевают, но все же остаются более сильными, и, даже если ослабевают, обычно сохраняют силу, достаточную для печати на клавиатуре компьютера и для выполнения других базовых функций современной жизни.
Дети с СМА типа 2 получают большую выгоду от физиотерапии и применения различных вспомогательных приспособлений всех видов. Приспособления для стояния и хождения, такие как легкие фиксаторы (ортезы) и ходунки, дают бОльшую мобильность, чем это было возможно в прошлом.
Многие дети способны управлять электроколясками или другими средствами передвижения довольно рано, уже в возрасте 3 лет или около того в зависимости от степени развития. Многие специалисты отмечают, что дети с СМА необычайно умны и сообразительны, а немногочисленные исследования подтверждают эти наблюдения
Наибольшую опасность при этом типе СМА представляет слабость дыхательных мышц. На протяжении всей жизни необходимо пристальное внимание к дыхательной функции и немедленное реагирование на инфекции. Ваш врач может помочь вам с особыми методами поддержания здоровья дыхательной системы, включая очистку дыхательных путей от выделений и возможную вентиляцию легких. Подробнее об этом см. «Слабость дыхательной мускулатуры»
Другая серьезна проблема при этом типе СМА — искривление позвоночника, обычно — в поперечном направлении, именуемое сколиозом. Сколиоз развивается из-за слабости мышц, поддерживающих позвоночник, который является гибким столбом. Сколиоз причиняет большие неудобства, препятствуя сохранению нормального положения тела и мобильности, и серьезно портя вид тела ребенка (или взрослого) Некоторые исследования показывают также, что тяжелое искривление позвоночника препятствует и дыханию
У многих детей с СМА сколиотическое искривление проявляется уже в начале жизни, и как правило корректируется с помощью фиксаторов до тех пор, пока не наступит подходящее время для хирургического вмешательства. Хирурги обычно склонны подождать, пока рост полностью (или почти) не прекратится, прежде чем хирургическим путем выправить и закрепить позвоночник. Они также наблюдают за дыхательной функцией ребенка и за тем, как быстро идет прогресс искривления
В настоящее время продолжительность жизни при СМА с дебютом в детстве варьируется. Вполне вероятно выживание до юношеского возраста и даже дольше
СМА тип 3 (болезнь Кугельберга-Веландер или умеренная СМА)
Некоторые источники описывают СМА тип 3 как СМА, начинающуюся в возрасте старше 18 месяцев, в то время как другие предпочитают говорить о ней как о СМА, начинающуюся после того, как ребенок начал ходить, или по крайней мере сделал самостоятельно пять шагов
Многие люди с этим типом СМА способны ходить до 30-40 лет, хотя некоторые теряют способность к хождению уже в юности
Важным остается пристальное внимание к дыханию и потенциальному искривлению позвоночника. При этом типе СМА обычно важны вспомогательные приспособления, такие как электроколяски, устройства для пользования компьютером и т. п. Некоторые люди обходятся только тростью и, иногда, складными стульчиками, а колясками пользуются только для передвижения на бОльшие расстояния, например в аэропорту или торговом центре
Люди с этим типом СМА доживают до зрелого возраста и достигают успехов в научной и трудовой деятельности
СМА тип 4 (СМА с дебютом во взрослом возрасте)
Этот тип СМА остается мягким на всем своем протяжении. По определению, заболевание при этом типе дебютирует в зрелом возрасте. Некоторые специалисты объединяют вместе типы 3 и 4, или типы 2 и 3
ЧТО ПРОИСХОДИТ С ЛЮДЬМИ С ДРУГИМИ ФОРМАМИ СМА?
Другие типы СМА, не связанные с дефицитом протеина SMN, значительно отличаются по тяжести и наиболее пораженным мышцам. При некоторых формах СМА, подобных SMN-связанным типам, наиболее поражены проксимальные мышцы, в то время как при других — наоборот дистальные, т. е. те, которые наиболее удалены от центра тела, по крайней мере в начале. На ранней стадии как правило поражаются кисти рук и стопы
Иногда у взрослых с заболеванием неясного происхождения, поражающим только мотонейроны спинного мозга и нижней части головного мозга, состояние относят к прогрессирующей мышечной атрофии. Это состояние иногда прогрессирует с вовлечением мотонейронов верхней части головного мозга, и тогда это состояние определяется как боковой амиотрофический склероз (БАС) Трансформация этого состояния в БАС происходит не всегда
Особенности спино-бульбарной мышечной атрофии (СБМА) рассмотрены ниже
Хотя все известные формы СМА — генетические, они являются результатом дефектов в различных генах и имеют разные типы наследования и прогнозы при планировании семьи
Если вам или вашему ребенку поставлен диагноз СМА, но не SMN-связанный тип, вам необходимо поговорить с вашим врачом, а возможно — и с генетиком-консультантом, чтобы больше узнать о генетике и прогнозах при этом определенном типе СМА
КАК СМА ЛЕЧИТСЯ?
Наибольшие потенциальные проблемы при СМА, особенно при формах, связанных с 5 хромосомой, в приблизительном порядке по серьезности, это:
слабость дыхательных мышцслабость глотательных мышцслабость мышц спины с прогрессирующим искривлением позвоночникааномальная реакция на препараты-миорелаксанты
Эти проблемы можно, и необходимо, лечить и предупреждать
Слабость дыхательных мышц
При наиболее тяжелых формах SMN-связанных СМА, и при некоторых других формах, слабость дыхательных мышц является серьезной проблемой. Это основная причина гибели детей с СМА типа 1 и 2. Когда дыхательные мышцы ослаблены, воздух не может хорошо циркулировать в легких, что негативно сказывается на общем здоровье. Симптомами слабости дыхательной мускулатуры являются головная боль, трудности со сном в ночное время, чрезмерная сонливость в дневное время, плохая концентрация, инфекции грудной клетки и, в конечном итоге, сердечная и дыхательная недостаточность
При СМА тип 1 очень ослаблены межреберные мышцы, в то время как мышцы диафрагмы остаются довольно сильными. В этом случае дети вынуждены дышать больше с помощью движений живота, чем грудной клетки, что придает их телу грушевидную форму
Родители новорожденных с СМА 1 типа могут столкнуться с вопросом, как продлить жизнь своего ребенка дольше, чем предполагаемые при этом типе два года. В последние годы доступность портативных и эффективных устройств для вентиляции легких дает родителям больше возможностей для этого. Некоторые такие дети к удивлению своих семей и докторов живут много лет, иногда — до подросткового возраста
Различные типы устройств вентиляции могут помочь взрослым и детям с менее тяжелым поражением. Многие специалисты рекомендуют начинать с неинвазивной вентиляции, которая как правило означает, что воздух (обычно комнатный воздух, не обогащенный кислородом) подается под давлением через маску или мундштук
Системы такого типа поставляются во многих формах и могут использоваться при необходимости по многу часов днем и/или ночью. Они легко могут быть сняты для еды, питья, разговора и, когда это возможно, для дыхания без их помощи. Некоторые пациенты предпочитают использовать вентиляционные системы с отрицательным давлением (разрежением), которые создают периодический вакуум вокруг грудной клетки или туловища, что помогает легким расширятся и сжиматься. Эти устройства работают по тому же принципу, что и применявшиеся несколько десятилетий назад т. н. «железные легкие», но при этом существенно менее громоздки
Для детей и взрослых с тяжелым поражением часто рекомендуется вентиляция через трахеостому — сделанное хирургическим путем отверстие в трахее, или дыхательном горле. В этом случае воздух под давление подается по трубке в трахеостоме. Обычно люди могут есть, пить и разговаривать с трубкой в трахеостоме, хотя этот процесс и требует некоторой адаптации. Существует некоторое беспокойство, что если ребенку делают трахеостому до того, как он начал говорить, это может вызвать сложности с его обучением речи
Другими важными аспектами решения дыхательных проблем при СМА является очистка дыхательных путей, для которой также иногда применяются механические устройства, и предотвращение инфекций столько, сколько это возможно
Слабость глотательных мышц
Слабость мышц рта и глотки вызывает проблемы с глотанием, особенно при наиболее тяжелых формах СМА. Дети со СМА тип 1 обычно испытывают трудности с глотанием и сосанием, что впоследствии приводит к их гибели. Слабость сосания приводит к обезвоживанию и недостаточному питанию, а трудности с глотанием — к непроходимости дыхательных путей вследствие вдыхания пищи или жидкости (это называется аспирац
sma-family.livejournal.com
 Генетическое заболевание Верднига-Гофмана относится к группе спинальных амиотрофий, наследуется по аутосомно-рецессивному типу.
Генетическое заболевание Верднига-Гофмана относится к группе спинальных амиотрофий, наследуется по аутосомно-рецессивному типу.
Спинальная мышечная атрофия (СМА) характеризуется врожденными или приобретенными дегенеративными изменениями в поперечнополосатых мышцах, симметричной мышечной слабостью туловища, конечностей, отсутствием или снижением сухожильных рефлексов при сохранении чувствительности.
Морфологические исследования выявляют патологию двигательных нейронов спинного мозга, «пучковую атрофию» в скелетных мышцах с характерным чередованием пораженных волокон и здоровых.
Отмечается нарушение проводящей функции нервных волокон, снижение сократительной способности мышц.Статистика
1 из 40-50 человек является носителем мутантного гена SMN. Проявляется патология с частотой 1 : 6 000 — 10 000 новорожденных.
Основной причиной спинальной амиотрофии Верднига Гоффмана является мутация гена SMN (от англ. survival motor neuron). Располагается ген выживания мотонейрона на 5 хромосоме, представлен двумя копиями:
Продуктом этого гена является белок SMN, участвующий в образовании и регенерации РНК.
Нехватка белка вызывает патологии двигательного нейрона.
В 95% случаев болезни Верднига-Гофмана наблюдается делеция (выпадение) SMNt, что вызывает дефицит белка SMN. Копия SMNc лишь частично компенсирует отсутствие теломерной копии.
Количество копий SMNc составляет от 1 до 5. Чем больше число центромерных копий, тем полнее воспроизводится белок и менее выражена патология нейрона.
Кроме количества копий SMNc, тяжесть заболевания определяется длиной участка делеции и генными конверсиями еще 3 генов: NAIP, h5F5, GTF2h3. Участием дополнительных модифицирующих факторов объясняется клиническое разнообразие симптомов.
Выделяю такие виды:
СМА 1 и СМА 2 имеет разные симптомы и признаки.
Первые симптомы выявляют еще во время беременности по слабому шевелению плода.

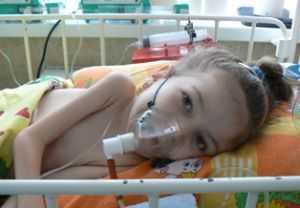
Фото: спинальная амиотрофия Верднига Гофмана
С самого рождения у детей наблюдается дыхательная недостаточность, врожденная спинальная амиотрофия Верднига Гофмана отмечаются:
Малыш принимает характерную позу «лягушки» с согнутыми в суставах руками и ногами, лежа на животе. При СМА 1 нередко отмечают частичный паралич диафрагмы – синдром Кофферата.
Явление характеризуется затруднением дыхания, одышкой, цианозом.
На стороне паралича наблюдается выбухание грудной клетки, повышается риск пневмонии.
У младенцев наблюдаются деформации костной системы, выражающиеся в ограничении подвижности суставов, появлении сколиоза, изменении формы грудной клетки.
Первые месяцы жизни дети развиваются нормально: вовремя начинают держать головку, сидеть, стоять.
После 6 месяцев появляются первые симптомы, обычно после острой респираторной или пищевой инфекции.
В первую очередь страдают конечности, особенно ноги, снижаются сухожильные рефлексы.
Затем в процесс постепенно вовлекаются мышцы туловища и рук, межреберные мышцы, диафрагма, что вызывает деформацию грудной клетки. Изменяется походка, приобретая сходство с «заводной куклой».
Дети становятся неловкими, часто падают. Наблюдаются подергивания языка, дрожание пальцев.
СМА 1 характеризуется злокачественным течением. Тяжелые расстройства дыхательной функции, сердечно-сосудистая недостаточность нередко приводят к смерти в первые месяцы жизни. До 5 лет доживают 12% больных.
СМА 2 также имеет тяжелый прогноз, хотя и протекает несколько мягче. Летальный исход отмечается в 14-15 лет.
 При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При обнаружении делеции теломерной копии SMNt диагноз считают подтвержденным.
В случае отсутствия делеции проводят дополнительные исследования:
При нормальных показателях фермента креатинкиназы проводят подсчет копий SMNc. В случае единственной копии идентифицируют точечную мутацию, принимая окончательное решение.
Похожие симптомы наблюдаются при врожденной миопатии – нарушении тонуса мышц.
Полностью исключить мышечную гипотонию позволяют результаты биопсии.
Определенное сходство с заболеванием Верднига-Гофмана имеет острый полиомиелит. Он начинается бурно, с резкого подъема температуры, несимметричных множественных параличей.
Несколько дней длится острый период, затем процесс переходит в восстановительную стадию.
Гликогенозы и врожденные миопатии также характеризуются сниженным мышечным тонусом. Изменения вызываются, в отличие от спинальной мышечной амиотрофии, нарушением обмена веществ, карциномой, гормональным дисбалансом. Следует исключить также болезнь Гоше, синдром Дауна, ботулизм.
Лечение спинальной амиотрофии симптоматическое и направлено на стабилизацию состояния пациента.
Назначают лекарственные средства:

Больным предписывают ортопедические процедуры в сочетании с теплыми ваннами, показаны лечебная гимнастика, мягкий массаж, оксигенотерапия, сульфидные ванны.
Условно различают проксимальные и дистальные формы СМА. 80% всех видов спинальных амиотрофий относятся к проксимальной форме.
К ним относятся, кроме заболевания Верднига-Гофмана:
В эту группу входит также болезнь Нормана, СМА с врожденным артрогрипозом, СМА с врожденными переломами.
К дистальным спинальным амиотрофиям относится прогрессирующий детский паралич Фацио-Лонде, болезнь Брауна-Виалетта-ван Лэре, СМА с параличом диафрагмы, эпилепсией и глазодвигательными нарушениями.
neurodoc.ru
Что предпринять, когда твоему ребенку ставят смертельный диагноз (СМА) и помощи нет ни от государства, ни от фондов
 День рождения Златы. 2 года.
День рождения Златы. 2 года.Родственники 2-летней Златы ОВЕРЧУК не каждый день, но регулярно вывозят девочку гулять. Парки, скверы, ровные площадки в горах – любые удобные для передвижения с коляской места. Поэтому, когда мы договариваемся об интервью, они готовы подъехать в сквер недалеко от редакции. Чтобы выгрузить из машины специализированную коляску, откашливатель и портативный аппарат искусственной вентиляции легких (ИВЛ) с пульсоксиметром, который показывает сердцебиение и насыщенность кислородом, а потом положить и подключить дочку, у мамы Надежды Яценко уходит минут 10–15.
Услышав историю девочки с диагнозом СМА – спинальная мышечная атрофия, кажется, что большинство врачей и чиновников, к которым с декабря 2016 года ее родные обращались за помощью, поступали точно так же. Ускоряли свой шаг. И не помогали. При этом диагнозе у человека с рождения прогрессируют мышечная слабость и атрофия мышечных волокон вследствие поражения мотонейронов (двигательных нервных клеток) в спинном или головном мозге. При этом заболевании каждый второй ребенок не доживает до 2 лет. То, что Злате удалось перешагнуть этот рубеж, вероятно, заслуга родственников и организованного ими качественного ухода. От государства их семье только 2 раза выдали бесплатные памперсы. А недавно из Министерства здравоохранения пришел ответ, что единственная медпомощь, на которую они могут рассчитывать, – лечебная физкультура.
Лечение от СМА существует. В конце сентября 2017 года первой страной в мире, где лечат своих граждан бесплатно, стала Италия. Лекарство было изобретено в 2015 году, в 2016-м оно после завершения тестирования поступило в официальную продажу.
Лечение от СМА существует. В конце сентября 2017 года первой страной в мире, где лечат своих граждан бесплатно, стала Италия. Лекарство было изобретено в 2015 году, в 2016-м оно после завершения тестирования поступило в официальную продажу. Пройти лечение на коммерческой основе можно в Германии – в Университетской клинике Фрайбурга готовы принять Злату. Стоимость одной ампулы лекарства – почти 90 тысяч евро, стоимость курса лечения в первый год, а это 8 ампул, – 702 тысячи евро. Во второй понадобится еще 3–4 ампулы. Таким образом, более миллиона евро. На сегодня собрано 4000 евро. Странички в соцсетях и даже два сюжета в эфире КТК (10 апреля 2017 года – выпуск «Внутри я танцую» в программе «Главная редакция» и 26 сентября 2017 года – вечерние новости) не помогли. Ни одного поступления от незнакомых людей. Все перечисленное – от друзей и друзей друзей. Родные Златы – бухгалтер и архитектор – понимают, что сумма огромная, но поиски денег продолжают.
 На прогулке
На прогулкеКак нам диагностировали СМА
– Моя беременность протекала очень тяжело. С первых дней у меня был сильный токсикоз, давление повышалось все время, а с 6 месяцев – тромбофлебит. Мне приходилось принимать серьезные препараты, ставить себе уколы в живот, чтобы не было преждевременных родов. Из-за отслоения плаценты мне экстренно провели кесарево сечение.
Первой заподозрила, что у Златы СМА 1-го типа, врач-невролог Ирина Борисовна КРАВЧЕНКО. Она отправила нас сдавать множество анализов. В том числе Злата проходила МРТ головы под щадящим наркозом (убирают маску – ребенок сразу приходит в сознание).
Мы ходили по врачам. Один известный педиатр, прием у которого стоит 20 тысяч тенге, сказал, что у нас «вялый ребенок». Первой заподозрила, что у Златы СМА 1-го типа, врач-невролог Ирина Борисовна КРАВЧЕНКО. Она отправила нас сдавать множество анализов. В том числе Злата проходила МРТ головы под щадящим наркозом (убирают маску – ребенок сразу приходит в сознание). Так как ребенок совсем маленький, то везде нам отказывали. Согласилась только одна клиника. Процедура длительностью 10 минут стоила нам 68 тысяч тенге. Мы с мужем сдавали анализ ДНК: заболевание генетическое, оно проявляется, если оба родителя являются носителями гена.
Также мы проходили ЭНМГ – игольчатый анализ (обкалывание тела иголками, чтобы понять, куда доходят нервные импульсы). Злате тогда было 3 месяца, хотя обычно его делают не раньше 5. Небольшие импульсы были только на малых берцовых костях, все остальное – без реакции. Итак, в 4 месяца нам диагностировали СМА1. Я рассказываю так подробно не потому, что жалуюсь, а чтобы вы понимали, что протоколов диагностики СМА в Казахстане нет и родители весь путь проделывают по наитию.
– В 5 месяцев у Златы начался обструктивный бронхит. Когда нас увезли по «скорой», то она задыхалась и синела. Нас госпитализировали в одну палату с ребенком с диагнозом пневмония. Я думаю, что именно это привело к дальнейшим событиям. 3 дня нам ничего не кололи, не назначали, мокрота не отходила. Оказывается, ее надо было санировать. За 5 дней она спустилась в легкие: из-за неработающих мышц горла кашель и естественное отхаркивание мокроты невозможны. С самого начала и до реанимации мы лежали в общей палате, хотя обещали дать отдельную, так как знали наш диагноз. Через 3 дня рентген показал пневмонию. Нас начали лечить так же, как и условно здоровых детей. А на следующий день Злата попала в реанимацию. Врачи сказали мне, что такие дети долго не живут. Наверное, только тогда до меня дошло полное осознание, насколько опасен диагноз дочери. Самое страшное, что никто не знал, как это лечить.
Тогда же Злате поставили трахеостому. Если бы не бронхит и пневмония, с этим диагнозом можно жить с маской на НИВЛ-аппарате. А с этими болезнями дышать без дырочки в горле она больше не может. Также нам понадобился откашливатель. В Казахстане официально, с гарантией, его можно купить за 5,5 миллиона тенге. В России стоимость аппарата в 2 раза ниже.
Пока везли откашливатель, пневмония стала двусторонней, а также ребенку сломали носовую перегородку. Она с момента поступления в реанимацию дышала через интубационную трубочку в носу. Вероятно, при очередной замене трубки и сломали перегородку. Сейчас любые манипуляции можно делать только через левую ноздрю. Откашливатель выводит не только мокроту, но и выступает в качестве слюноотсоса – слюноотделение у детей с СМА нарушено.
Также благодаря неравнодушным людям, Гульнар ДОСАЕВОЙ и семье Ольги и Евгения КОКОРКИНЫХ, мы приобрели два портативных аппарата искусственной вентиляции легких. Второй появился, когда первый дал сбой и дыхание Златы в течение нескольких часов мы попеременно поддерживали с помощью мешка Амбу – ручного аппарата искусственной вентиляции легких.

– Текущие расходы, а это 300–350 тысяч тенге в месяц, по уходу за Златой наша семья покрывает сама. Например, аминокислотное питание, которое ей нужно, стоит 15 000 тенге за баночку. В Казахстане оно не продается. Поэтому мы всеми путями, оказиями ищем, как заказать и привезти его сюда. Также мы сами покупаем комплектующие и расходные материалы к двум аппаратам искусственной вентиляции легких. Они работают в круглосуточном режиме. Я проделываю манипуляции каждые 15–20 минут, ночью просыпаюсь 3–4 раза. Поэтому расходников требуется очень много. Я по внешнему виду дочки, по ее дыханию и давлению (аппарат показывает, что сатурация падает) определяю, что ей нужно прочистить легкие от мокроты.
Когда мы узнали о существовании лекарства, понимали, что оно будет дорогое. Думали, 10–20 тысяч евро. Но когда узнали, что 90 тысяч евро, а инъекций за 2 года надо 12, то были в шоке. Для нас это астрономическая сумма!
Один из эффектов лекарства, на которое мы собираем деньги, – сокращение выделения мокроты. Также оно неким образом восполняет дефицит белка, отсутствие которого приводит к атрофии мышечных волокон. У детей его можно применять с рождения. На Западе СМА умеют диагностировать сразу. На испытания брали детей в возрасте до 7 месяцев – к 2 годам они самостоятельно ели и ходили. Когда мы узнали о существовании лекарства, понимали, что оно будет дорогое. Думали, 10–20 тысяч евро. Но когда узнали, что 90 тысяч евро, а инъекций за 2 года надо 12, то были в шоке. Для нас это астрономическая сумма!
Пока что мы собираем деньги на первый год лечения (702 000 евро) с надеждой, что лекарство подешевеет и станет доступно каким угодно образом нам, казахстанцам. Единственная загвоздка в том, что на экспериментальную терапию почти не брали детей с трахеостомой. Но мы надеемся, что со временем Злата покажет хорошие результаты, научится сама дышать, держать головку, есть, двигать ножками и ручками и, конечно, стоять и ходить. На все воля Бога. Но пока все решают деньги, которых у нас нет. А время бежит очень быстро.
Официальной статистики, сколько детей с СМА, в Казахстане нет. По нашим данным, а мы узнаем в основном уже об ушедших детях, за последние 1,5 года ушло 5 детей: 4 со СМА1, 1 мальчик со СМА2. Есть еще недиагностированные случаи: у одной мамы первый ребенок умер по неизвестной причине в возрасте 7 месяцев, второму малышу, рожденному позже, уже поставили диагноз. Протоколов лечения и оказания помощи таким детям в Казахстане тоже нет. В сентябре пришло письмо, где вместо медицинской помощи нам рекомендовано ЛФК. Мы считаем это издевкой. Девочке, у которой вовсе не движутся руки и ноги, рекомендовать ЛФК! Мы узнаем некоторые вещи через российский интернет-форум семей со СМА, на сайтах smanewstoday.com и spinraza.com. Мы бережно переводим все материалы, ссылки на которые там публикуют, чтобы понимать, что происходит с ребенком.
Вероятность рождения ребенка со СМА у родителей-носителей генов – 25%. Сейчас существует анализ, способный внутриутробно выявить болезнь. Но я не решусь заводить еще одного ребенка. Мы православные. Я не смогу сделать аборт, если СМА подтвердится. А жизнь с таким заболеванием сегодня – это то, чего ни одна мать своему ребенку не пожелает.
У всех, и у благотворительных фондов, и у простых людей, услышавших о нашей беде, один вопрос: «Это полностью излечивается?» А что мы можем сказать, если препарат экспериментально применяется 2 года? Есть значительные улучшения, а как оно будет дальше – неизвестно. Для родителей это же не повод отказаться от веры в то, что их ребенок будет жить.
 Факты про СМА:
Факты про СМА:По данным Минздрава США, СМА диагностируют каждый год у 1 из 10 000 новорожденных. Такие крошечные группы пациентов называют «сиротскими».
Цена является «возмутительно высокой» (формулировка журналиста The Washington Post) даже для американцев. Там препарат стоит 125 тысяч долларов. Покрыть расходы на препарат не могут и страховые компании. Шанс родителей на лечение появляется, когда они подписывают соглашение с фармацевтической компанией: лечение проводится в обмен на участие в маркетинговых активностях. Итальянские семьи, принимающие участие в экспериментальном лечении, также обязаны регулярно выкладывать материалы о динамике лечения.
Для новорожденных с СМА1 продолжительное выживание стало возможным с помощью поддержки питания и вентиляции легких, но не улучшило моторное развитие. У пациентов с менее тяжелыми типами заболеваний уровень ухода за ними влияет на замедление прогрессирование заболевания.
Для пациентов со СМА дома может понадобиться набор аппаратуры из 7 единиц. Необходимый минимум: пульсоксиметр, аппарат искусственной вентиляции легких, отсасыватель мокроты, кислородный концентратор.
Летом 2017 года российские издания писали о 28-летнем мужчине, страдающем СМА и неспособном двигаться, который был признан судом виновным в вооруженном разбое по делу о похищении мотороллера. Суд назначил инвалиду, который практически полностью парализован и весит около 20 килограммов, наказание в виде четырех с половиной лет колонии. Мужчина имеет высшее образование и семью, занимается благотворительностью. После вмешательства СМИ и правозащитников его освободили.
Контактный телефон:
8 702 201-31-23 Надежда ЯЦЕНКО (мама)
Маргарита ЮРЧЕНКО (тетя)СБЕРБАНК РОССИЯПолучатель Юрченко Маргарита Викторовна№ счета 40820810738257007049БИК 044525225Кор. счет 30101810400000000225КПП 773601001ИНН 7707083893ОКПО 57972160ОГРН 1027700132195
Мегафон +7 925 934-50-64
QIWI-кошелек +7 777 270-99-28
АО «Казкоммерцбанк»карта № 5483 1828 4596 3538, KZ97926020P555005244Яценко Надежда, ИИН 830 308 400 935
АО «АТФБанк»№ карты 4052 5660 0023 8922, KZ89826A1KZTW4013367Юрченко Маргарита, ИИН 780 224 403 298
The Bank of New York Mellon,SWIFT IRVTUS3N New York, USACor.acc.no 890 0260 645ATFBank Branch AlmatySWIFT ALMNKZKAacc.no KZ47826A1USDD3054532 (USD) / Yatsenko Nadezhda, Almaty, Kazakhstan
zoj.kz
Если семейный анамнез не отягощен, то есть в семье ни у кого не было подобного заболевания, то предсказать СМА невозможно. Поскольку оно рецессивное и относится к редким заболеваниям, в скрининг, как правило, берется набор других заболевании, и СМА туда не входит. Во многих странах пробовали проводить такой скрининг, но он оказался нерентабельный, и скрининг на все генетические заболевания провести невозможно.
Чтобы диагностировать СМА, нужно:
Клиническое обследование: при подозрении на СМА – консультация невролога и генетика.
Биохимия крови: креатинкиназа – в норме при СМАI, в норме или незначительно повышена при других типах.
Генетическое обследование: пренатально или постнатально. Тест ДНК путем определения делеции гена SMN1. Рекомендуется иметь результат и на количество копий гена SMN2. Этот анализ нужен для участия в клинических исследованиях.
Электронейромиография: показывает поражение мотонейронов спинного мозга (ритм-частокола или гигантские ПДЕ), помогает дифференцировать СМА от других нервно-мышечных болезней. Проводимость по периферическим нервам обычно нормальная.
Биопсия мышц: определяет гистологические признаки атрофии мышечных волокон, помогает дифференцировать СМА от других нервно-мышечных болезней.
Дифференциальная диагностика: определяет болезни мотонейрона, первичный боковой амиосклероз, мышечную дистрофию другие заболевания.
У людей старшего возраста СМА может быть похожа на БАС, но чаще всего СМА путают с миодистрофией Дюшенна. Возможно, это связано с тем, что в свое время много ведущих российских ученых занимались миодистрофией Дюшенна, было ряд публикаций в журналах, и у врачей это заболевание на слуху. Поэтому любое двигательное нарушение в раннем детском возрасте врачи обычно относят к миодистрофии Дюшенна. Эти заболевания часто путают еще и потому, что них развивается проксимальная мышечная слабость, хотя неврологически есть очень четкий критерий, и их можно отличить. Но бывают очень сложные случаи, когда трудно поставить правильный диагноз, поэтому необходимо обращаться в специализированные центры.
Неправильный диагноз при генетически детерминированных заболеваниях может привести к неправильному прогнозу на рождение в семье повторного больного. Для пациентов со СМА и с миодистрофией Дюшенна прогноз разный.

http://www.f-sma.ru/uploads/files/library/SMA_primary_diagnistics.pdf
spravka.neinvalid.ru